IVDR: wichtige Überlegungen zur Validierung eines Durchflusszytometrie-Assays
nach den Anforderungen der ISO 15189
Brice Ezzouaouy, Global Product Manager | Beckman Coulter Life Sciences, Marseille (France)
THEMENÜBERBLICK:
- Bedeutung der ISO 15189 für laborentwickelte Durchflusszytometrie- Tests, insbesondere im Kontext der neuen EU‑Verordnung 2017/746 über In‑vitro-Diagnostika (IVDR)
- Wichtigste Assay-Parameter, die verifiziert oder validiert werden müssen, um die Anforderungen der ISO 15189 zu erfüllen
- Technische Tipps und konkrete Methoden zur Erfüllung der Anforderungen der ISO 15189
Einleitung
Die ISO 15189:2012, „Medizinische Laboratorien – Anforderungen an die Qualität und Kompetenz“ (ISO 15189), definiert Anforderungen an die Qualität und Kompetenz medizinischer Labore. Wie andere ISO-Normen, die der globalen Standardisierung von Praktiken dienen, hat die ISO 15189 zum Ziel, die auf das Qualitätsmanagementsystem (QMS) bezogenen Anforderungen für medizinische Labore, die In‑vitro-Diagnostik durchführen, zu vereinheitlichen.
Während bislang nur wenige Länder der Welt eine Akkreditierung nach der ISO 15189 vorschreiben, wird sie mit der anstehenden EU-Verordnung 2017/746 über In‑vitro-Diagnostika (IVDR) für alle klinischen Labore in der Europäischen Union, die laborentwickelte Tests (LDTs) durchführen, verpflichtend. LDTs spielen insbesondere in Fachgebieten wie der Durchflusszytometrie eine wichtige Rolle, da nicht für alle diagnostischen Anwendungen gebrauchsfertige IVDLösungen im Handel erhältlich sind. Dies gilt besonders für die Hämatoonkologie.
Die ISO-15189-Akkreditierung eines Durchflusszytometrie-LDT ist alles andere als trivial und erfordert eine sorgfältige Planung. Unter den vielen Anforderungen der ISO 15189 kann sich vor allem die Leistungsvalidierung des Assays schwierig gestalten, da die Norm weder ein Standardverfahren hierfür noch nähere Angaben darüber enthält, welche Versuche durchgeführt werden sollten. In diesem Whitepaper beleuchten wir einige der wichtigsten Leistungsmerkmale, die nach der ISO 15189 validiert werden müssen, die speziell Durchflusszytometrie-Labore betreffenden Herausforderungen hierbei und einige wichtige Überlegungen hinsichtlich einer erfolgreichen Erlangung der ISO‑15189-Akkreditierung.
ISO 15189: Überblick und Zielsetzungen
Die auf die Normen ISO 17025 und ISO 9001 aufbauende ISO 15189 ist die einzige globale Norm für die Akkreditierung von medizinischen Laboren und legt die Anforderungen an deren Kompetenz und Qualitätssysteme fest. Die länderübergreifende Harmonisierung und Standardisierung der Qualität und Kompetenz von medizinischen Laboren mittels einer Norm ist zweifelsohne sinnvoll. Schätzungen zufolge ist die Labormedizin an 70 Prozent aller klinischen Entscheidungen beteiligt. Allein im Vereinigten Königreich werden pro Einwohner und Jahr durchschnittlich 14 Tests1
von einem Facharzt für Labormedizin durchgeführt. Es ist daher von entscheidender Bedeutung, dass IVD-Tests unter kontrollierten Bedingungen stattfinden.
Die Norm ISO 15189 ist sehr umfassend und deckt das gesamte QMS des Labors ab. Sie beinhaltet mehr als 170 Artikel, zuzüglich Anhängen, und empfiehlt, die Mitarbeiter auf allen Ebenen vollständig einzubinden und ihre Fähigkeiten zu nutzen, um die Organisation zu verbessern. Ziel ist es, dass die Mitarbeiter genau wissen, was zu tun ist, wie sie dabei vorgehen müssen, wer für einen Prozess verantwortlich ist und wo sämtliche für die eigene Tätigkeit notwendigen Informationen zu finden sind.
Der Inhalt der Norm gliedert sich hauptsächlich in Anforderungen an das Management (Teil 4) und technische Anforderungen (Teil 5).
Tabelle 1: Inhalt der Teile 4 und 5 der ISO 1518922
|
|
|
|
Nach ihrer Erstveröffentlichung vor fast zwei Jahrzehnten (2003) wurde die ISO 15189 im Jahr 2007 zur Angleichung an die ISO/IEC 170253 sowie nochmals 2012 überarbeitet. Obwohl sie einen Übergang von länderspezifischen Anforderungen zu einer international anerkannten Norm einleiten sollte, hat sie bislang noch keine große Verbreitung gefunden. Sie wird zwar mittlerweile in mehr Ländern angewendet, ist jedoch Stand 2020 weder in einigen größeren europäischen Ländern noch in den USA verpflichtend.
Ein Grund, weshalb sie nicht weltweit auf breiter Front eingeführt wurde, ist ihre komplexe Umsetzung. Die Anwendung der ISO 15189 erfordert viel Geld, Zeit und Ressourcen und ist kein einmaliger Aufwand, sondern ein stetiger Weg, da der Grundsatz der Norm die ständige Verbesserung ist. Die kleinsten Labore verfügen in der Regel nicht über die Kapazität, ein solch umfassendes QMS umzusetzen. Dies lässt sich sehr gut am Beispiel Frankreichs illustrieren, das zu den ersten Ländern gehörte, in denen die ISO 15189 verpflichtend wurde (ab 2011). Dort hat sich die Landschaft der klinischen Labore im Lauf von 10 Jahren völlig verwandelt: Gab es im Jahr 2008 noch 5000 Labore, waren es ein Jahrzehnt später nur noch 900. Diese Umstrukturierung wird vor allem darauf zurückgeführt, dass kleinere Labore konsolidieren mussten, um die Akkreditierungsanforderungen erfüllen zu können.4
Die durchschnittlichen Kosten der Akkreditierung betragen für ein Labor laut Schätzungen in der Anfangsphase 445 000 € und im Weiteren 145 000 € pro Jahr.5
Eine weitere Herausforderung, der sich Labore bei der Umsetzung der ISO 15189 gegenübersehen, ergibt sich dadurch, dass in der Norm nicht angegeben ist, welche Versuche und spezifischen Protokolle durchgeführt werden sollten, um die technischen Anforderungen zu erfüllen. Dadurch bleibt ein Interpretationsspielraum, der es erschwert, eine abstrakte Anforderung in einen konkreten Aktionsplan zu übersetzen.
ISO 15189: die tragende Säule für IVDR-konforme LDTs
Die neue EU-Verordnung 2017/746 über In‑vitro-Diagnostika (IVDR) ist am 26. Mai 2017 in Kraft getreten und sieht eine Übergangsfrist von 5 Jahren vor. Die IVDR ersetzt die EU-Richtlinie (98/79/EG), durch die In‑vitro-Diagnostika (IVD) seit 1993 geregelt wurden. Anlass hierzu waren Schwierigkeiten, die bei der Interpretation und Anwendung der Richtlinie auftraten und vor allem mit der schwachen Kontrolle von IVD mit potenziell „hohem Risiko“ zusammenhingen.
Für IVD-Hersteller bedeutet die IVDR eine Revolution. Während unter der EU-Richtlinie (98/79/EG) die meisten CE‑IVDProdukte eigenzertifiziert wurden, benötigen unter der IVDR nun bis zu 90 % der Branche eine Benannte Stelle, und die Hersteller müssen zusätzliche (und erhebliche) Anforderungen in Bezug auf ihr Qualitätsmanagementsystem (QMS) erfüllen – die alle relevanten Bereiche berühren, von der Assay-Leistungsvalidierung bis zur proaktiven Überwachung nach dem Inverkehrbringen.
Die IVDR betrifft nicht nur IVD-Hersteller, sondern die gesamte Landschaft der klinischen Diagnostik einschließlich der Endanwender. Dies gilt besonders für klinische Labore, die mit laborentwickelten Tests (LDTs) arbeiten. Die IVDR erkennt zwar an, dass zur Diagnose bestimmter Erkrankungen LDTs benötigt werden, sieht aber gleichzeitig das Risiko, das zu wenig kontrollierte Hochrisiko-LDTs bergen. Wenn die Messlatte für Hersteller höher gelegt wird, ist es nur logisch, dass auch in Laboren entwickelte und hergestellte IVD-Tests entsprechend reguliert werden.
Um LDTs nutzen zu können, müssen klinische Labore nun verschiedene neue Anforderungen erfüllen, die größtenteils in Artikel 5 (5) der IVDR ausgeführt sind. Labore, die LDTs durchführen, doch die Anforderungen des Artikels 5 (5) nicht erfüllen, werden IVD-Herstellern gleichgestellt, das heißt, sie müssen die gesamte Verordnung einhalten (z. B. klinische Nachweise erbringen, eine Benannte Stelle beauftragen und Prozesse für die Überwachung nach dem Inverkehrbringen einrichten). Da es schon IVD-Herstellern schwerfällt, diese Anforderungen zu erfüllen, obwohl sie über große, funktionsübergreifende Teams verfügen, die die Entwicklung und Vermarktung von Tests für EU-Labore betreuen, bleibt abzuwarten, wie viele Labore die Kapazität haben, LDTs durchzuführen, ohne die Anforderungen des Artikels 5 (5) zu erfüllen.
Eine zentrale Forderung des Artikels 5 (5) besteht darin, dass Labore die Norm ISO 15189 oder ggf. entsprechende nationale Vorschriften, einschließlich nationaler Akkreditierungsregeln, erfüllen müssen.
Nachstehend sind die typischen Abläufe in einem klinischen Labor bei der Validierung von CE-IVD-Assays bzw. LDTs im Kontext der IVDR den entsprechenden Abläufen unter der IVDD gegenübergestellt. Im Bereich der Durchflusszytometrie könnten Panels unter Verwendung individueller CE-zertifizierter konjugierter Antikörper den Validierungsablauf deutlich verbessern, da der Assay in diesem Fall nicht als LDT eingestuft wird und das Labor somit möglicherweise nicht die Anforderungen des Artikels 5 (5) erfüllen muss. ISO‑15189-Konformität oder ggf. andere nationale Akkreditierungsvorschriften sind eine tragende Säule des Validierungsprozesses im Rahmen der IVDR und insbesondere für die Leistungsvalidierung von LDTs.
Abbildung 1: Gegenüberstellung der Validierung von IVD-Tests und LDTs im Kontext der IVDD und der IVDR
 Infografik IVDD vs. IVDR ansehen
Infografik IVDD vs. IVDR ansehen
LDT-Leistungsvalidierung nach ISO 15189: wichtige Überlegungen
Ein zentrales Element der ISO 15189 sind die technischen Anforderungen, die sicherstellen sollen, dass IVD-Assays eine ausreichende Leistung aufweisen. Die Methodenvalidierung bildet möglicherweise den schwierigsten und zeitaufwändigsten Teil der Umsetzung der ISO 15189. Die technischen Anforderungen sind in den in Tabelle 1 genannten Teilen 5.3, „Laboratoriumsausrüstung, Reagenzien und Verbrauchsgüter“, und 5.5, „Untersuchungsverfahren“, aufgeführt.
Die ISO 15189 enthält abstrakte Richtlinien, die Spielraum zur Anpassung des Akkreditierungsverfahrens an örtliche Gegebenheiten lassen. Dies macht die Sache für die medizinischen Labor nicht leichter, denn sie müssen bestimmen, welche Verfahren und Versuche sie durchführen müssen, um die Anforderungen zu erfüllen. Einige nationale Institutionen haben konkretere technische Leitfäden erstellt, um ihre Labore dabei zu unterstützen. Dies ist zum Beispiel in Frankreich der Fall, wo die nationale Akkreditierungsstelle COFRAC konkrete technische Empfehlungen und Hinweise zur Methodik herausgegeben hat.6 Ein Großteil der Inhalte dieses Whitepapers stützt sich auf die COFRAC-Empfehlungen. Das vorliegende Dokument ist keinesfalls ein erschöpfender Leitfaden und eine andere Vorgehensweise kann sicherlich akzeptabel sein, solange sie begründet und dokumentiert ist.
Methodenvalidierung – Prozessüberblick
Die Umsetzung der IVDR muss, insbesondere im Hinblick auf die Methodenvalidierung, sorgfältig geplant werden. In der Abbildung unten sind die wichtigsten Schritte zur Verifi zierung oder Validierung einer Methode dargestellt.
Abbildung 2: Die wichtigsten Schritte zur Verifi zierung oder Validierung einer Methode

Wenn das klinische Labor eine Methode einführt, die bereits validiert ist (z. B. Test mit CE-IVD-Kennzeichnung, der gemäß Herstelleranweisung und innerhalb der angegebenen Zweckbestimmung angewendet wird), muss es nur verifi zieren, dass die vom Hersteller angegebenen Leistungsmerkmale in der eigenen Laborumgebung erreicht werden. Wenn das klinische Labor eine intern entwickelte Methode einführt, oder eine validierte Methode an die eigenen Erfordernisse anpasst, muss es die Leistungsmerkmale validieren. In der nachstehenden Tabelle sind Beispiele für Parameter aufgeführt, die bei der Verifi zierung oder Validierung von quantitativen bzw. qualitativen Methoden beurteilt werden müssen.
Tabelle 2: Parameter, die bei quantitativen Methoden und bei qualitativen Methoden verifiziert oder bestimmt werden müssen6
Quantitative Methode
Qualitative Methode
Auf Grundlage der Ergebnisse muss das Labor die Eignung der Methode bestimmen. Wenn definierte Spezifikationen nicht erfüllt werden, muss das Labor deren Akzeptanz begründen.
Wiederholpräzision und Genauigkeit
Zwei maßgebliche, zu validierende Parameter sind die Wiederholpräzision und die Genauigkeit eines Systems. Sie sind voneinander unabhängig. Ein IVD-Test könnte genau, aber nicht wiederholbar sein, oder er könnte umgekehrt eine hohe Wiederholpräzision aufweisen, doch häufig das Ziel verfehlen, wie in Abbildung 3 dargestellt ist.
Abbildung 3: Wiederholpräzision und Genauigkeit im Vergleich

Zur Beurteilung der Wiederholpräzision wird dieselbe Probe innerhalb eines kurzen Zeitraums mehrfach analysiert – mit demselben Bediener, derselben Reagenzcharge und demselben Gerät. Es sollten möglichst 2 Konzentrationsstufen, davon vorzugsweise eine nahe des Cutoff -Werts für die Entscheidung, analysiert werden, und in der Regel sind 30 Tests ideal, um statistische Relevanz sicherzustellen. Die Anzahl der Tests hängt von etwaigen Limitationen ab: Probenverfügbarkeit, Kosten der Reagenzien, Dauer des Versuchs usw. Der Variationskoeffi zient wird aus dem Mittelwert und der Standardabweichung berechnet (= Standardabweichung / Mittelwert × 100) und muss niedriger als der vorab festgelegte Zielwert der Wiederholpräzision sein. Die Wiederholpräzision muss für jeden Probentyp (z. B. Vollblut, Knochenmark) beurteilt werden. Gegebenenfalls muss die Wiederholpräzision nach einer erheblichen Veränderung des Systems, beispielsweise nach Wartungsmaßnahmen, erneut beurteilt werden.
Die Beurteilung der Genauigkeit umfasst üblicherweise einen Vergleich mit einem Zielwert unter Verwendung von externen Qualitätskontrollen und/oder Ringversuchsprogrammen. Der Zielwert ist der Durchschnittswert aller Teilnehmer für einen standardisierten Parameter (z. B. Konsenswert) oder der Durchschnittswert der Teilnehmer, die dieselbe Methode anwenden. Der systematische Fehler des validierten Assays in Prozent wird wie folgt berechnet:
systematischer Fehler (%) = (Durchschnitt aus Reproduzierbarkeitsprüfung innerhalb des Labors – Zielwert) / Zielwert × 100.
Weitere Einzelheiten hierzu enthält die ISO 5725-4, „Genauigkeit (Richtigkeit und Präzision) von Messverfahren und Messergebnissen – Teil 4: Grundlegende Methoden für die Ermittlung der Richtigkeit eines vereinheitlichten Messverfahrens“.7
Zwischenpräzision (laborinterne Reproduzierbarkeit)
Bei der Beurteilung der Wiederholpräzision bleiben alle Parameter unverändert. Zur Berechnung der Zwischenpräzision (auch als laborinterne Reproduzierbarkeit bezeichnet) wird dieselbe Probe mehrfach unter verschiedenen Bedingungen (z. B. andere Bediener, Zeitpunkte, Reagenzchargen, Geräte) analysiert. Ein typisches Protokoll beinhaltet 30 Tests mit zwei Konzentrationen über 15 Tage. Andere Ansätze sind zulässig, sofern sie statistisch begründbar sind. Auch hierbei wird der Variationskoeffi zient aus dem Mittelwert und der Standardabweichung berechnet und muss niedriger als der vorab festgelegte Zielwert sein.
Linearität, Nachweisgrenze und Quantifizierungsgrenze
Die Nachweisgrenze ist das kleinste Signal, das noch vom Hintergrund unterschieden werden kann. Sie kann anhand von 30 Messungen an einer Negativkontrolle als das 3‑Fache der Standardabweichung definiert werden. Bei qualitativen Methoden entspricht die Nachweisgrenze der Positivschwelle.
Die Quantifizierungsgrenze ist der kleinste gemessene Wert innerhalb eines akzeptablen Konfidenzniveaus. Sie kann dem 10‑Fachen der Standardabweichung aus 30 Negativkontrollmessungen entsprechen. Alternativ kann sie wie folgt durch Verdünnung einer Kontrolle bestimmt werden:
- 11 Verdünnungen einer Kontrolle (z. B. 100 + 0; 90 + 10…10 + 90; 0 + 100).
- 10-malige Messung jeder Verdünnung und Bestimmung des Variationskoeffizienten.
- Die Quantifizierungsgrenze wird definiert als die niedrigste Konzentration, die einen VK < 10 % ergibt.
Als weitere Möglichkeit der Verdünnung können verschiedene Gemische von positiven und negativen Zelllinien beurteilt
werden.
Die Linearität zwischen den gemessenen Verdünnungen und Konzentrationen muss verifiziert werden. Die obere Linearitätsgrenze und die Nachweisgrenze müssen den zu erwartenden Konzentrationsbereich der Proben abdecken, die im Labor getestet werden.
Unsicherheit und Streuungsfaktoren
Bei jedem IVD-Assay muss unbedingt definiert werden, welche Faktoren das Ergebnis beeinflussen könnten und welche dieser Faktoren nicht signifikant sind (mit Begründung), und es muss nachgewiesen werden, dass die übrigen Faktoren kontrolliert werden. In der ISO 15189, Abschnitt 3.17, ist die Messunsicherheit definiert als „Parameter im Zusammenhang
mit dem Ergebnis einer Messung, der die Streuung der Werte charakterisiert“.
Zur Bewertung der Messunsicherheit gibt es nicht den einen, sondern mehrere Ansätze. So kann sie beispielsweise anhand der Ergebnisse von internen und externen Qualitätskontrollen definiert werden, indem die Ergebnisse als Wert ± U angegeben werden, wobei U die nach der folgenden Formel berechnete Unsicherheit ist: U = √ ((A2 + B2))
Wobei:
A = Varianz (SD2) aus allen internen QK
B = (Mittelwert aus Reproduzierbarkeitsstudie − Zielwert) × 100) / Zielwert
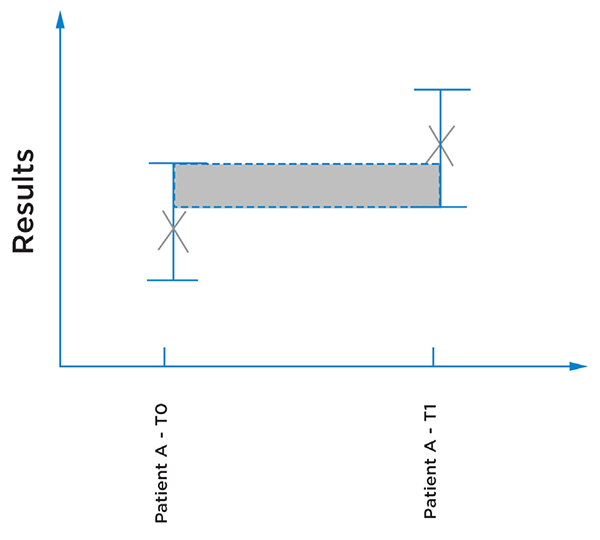
Abbildung 4: Darstellung der Patientenergebnisse von zwei verschiedenen Zeitpunkten; die Fehlerbalken zeigen die Unsicherheit der Ergebnisse an. In diesem Fall kann nicht darauf geschlossen werden, dass sich die Ergebnisse unterscheiden, da die Unsicherheit in der Ungenauigkeit der Messung inhärent sein könnte.
Reagenzstabilität
Die Bestimmung der Reagenzstabilität ist nur im Fall der Methodenvalidierung relevant, da sich das Labor ansonsten auf die vom Hersteller bereitgestellten Daten verlassen kann, solange es die Gebrauchsanweisung und die Zweckbestimmung der Reagenzien und die Lagerbedingungen für ungeöffnete und angebrochene Fläschchen einhält. Die Stabilität eines Reagenzes muss für das ungeöffnete Fläschchen (Haltbarkeit des Reagenzes) und das angebrochene Fläschchen nachgewiesen werden. Dies gilt besonders für in der Durchflusszytometrie eingesetzte konjugierte Antikörper, denn die Produktleistung kann durch mehrere Faktoren beeinträchtigt werden, wenn das Fläschchen angebrochen und in Verwendung ist (z. B. Oxidation, Abbau durch Licht).8
Ein klassischer Ansatz zur Prüfung der Reagenzstabilität ist das Testen einer Probe mit bekanntem Wert (z. B. einer Kontrolle) von Tag 1 bis Tag n mit mindestens 10 Replikaten zwischen Tag 1 und n. Die akzeptable Streuung des gemessenen Parameters wird vorher festgelegt (z. B. ± 10 %). Als Stabilitätsgrenze wird der Zeitpunkt des letzten Werts festgelegt, der noch in den akzeptablen Streuungsbereich fällt.
Abbildung 5: Darstellung der Ergebnisse einer Reagenzstabilitätsprüfung über 60 Tage. In diesem Beispiel beträgt die Stabilität des Reagenzes 50 Tage, denn nach diesem Zeitpunkt liegen die Ergebnisse außerhalb der akzeptablen Streuung (hier ± 10 %).

Methodenvergleich
Der Methodenvergleich erfolgt nach der Verifizierung anderer Kriterien (z. B. Wiederholpräzision, laborinterne Reproduzierbarkeit, Genauigkeit, Linearität), indem innerhalb kurzer Zeit mindestens 30 Proben im Konzentrationsbereich, der bei den Analysen im Labor zu erwarten ist, sowohl mit der zu validierenden Methode als auch mit der Referenzmethode analysiert werden. Abweichungen werden ausgewertet, um sicherzustellen, dass sie innerhalb vorab festgelegter Grenzen liegen. Wenn einige Proben scheitern, müssen die möglichen Ursachen ermittelt werden (z. B. Antikoagulans, Alter der Probe) und bei Bedarf Gegenmaßnahmen ergriffen werden.
Abbildung 6: Darstellung eines Vergleichsdiagramms, das die Übereinstimmung zwischen einer neuen Methode und der Referenzmethode, gegen die sie validiert wird, zeigt

Probenverschleppung
Die Probenverschleppung spielt vor allem bei automatisierten Systemen und sensiblen Parametern (z. B. Beta‑HCG) eine Rolle. Sie ist besonders wichtig für die Beurteilung von Probenvorbereitungssystemen und Reinigungs- bzw. Dekontaminationssystemen.
Ein Protokoll zur Beurteilung der Probenverschleppung könnte etwa die 3‑fache Analyse einer Probe mit hoher Konzentration (H1, H2, H3, Mittelwert mH) gefolgt von der 3‑fachen Analyse einer Probe mit niedriger Konzentration (B1, B2, B3) vorsehen. Diese Sequenz würde 5‑mal wiederholt, und es würden die Mittelwerte von B1 und B3 berechnet. Durch einen t‑Test kann das Labor bestimmen, ob sich mB1 von mB3 unterscheidet. Die Kontamination in Prozent kann wie folgt berechnet werden: (mB1 − mB3) / (mH − mB3) × 100.
Wenn für alle Reagenzien dasselbe Verteilungssystem verwendet wird, kann auch die Reagenzverschleppung beurteilt werden. Hierbei würde Parameter A 10‑mal an derselben Probe gemessen. Danach würde Parameter A erneut 10‑mal gemessen, jedoch abwechselnd mit Parameter B. Anhand eines t‑Tests würde dann bestimmt, ob sich die Differenz zwischen den beiden Mittelwerten bei Parameter A statistisch unterscheidet.
Validierungs-/Verifizierungsdossier und ständige Leistungsüberwachung
Datenerhebung ist eine gute Sache, aber offensichtlich nutzlos, wenn die Daten nicht entsprechend ausgewertet und zusammengefasst werden. Im Kontext der ISO‑15189-Akkreditierung muss das Labor für jeden Test ein Dossier erstellen. Jedes Dossier enthält mindestens Folgendes:
- Beschreibung des Analysenprozesses (zu verifizierende/validierende Schritte, Methoden, Elemente)
- Risikomanagement
- Bestimmung der zu beurteilenden Leistungskriterien
- Bestimmung der Spezifikationen oder Akzeptanzgrenzen für diese Kriterien
- Bibliographieverifizierung
- Versuchsplan und Implementierungsmethodik
- Zusammenstellung und statistische Analyse der erhobenen Daten
- Schlussfolgerung und Entscheidung bezüglich der Validität des Assays gemäß den festgelegten Leistungskriterien
Nationale Einrichtungen wie die COFRAC in Frankreich stellen Vorlagen für diese Dossiers bereit.9
Die tragende Säule der ISO 15189 ist die ständige Verbesserung. Die Leistungsvalidierung ist kein einmaliger Aufwand. Durch die Nutzung von statistischen Daten aus internen und externen Qualitätskontrollen können Labore die Validität der folgenden Leistungsmerkmale im zeitlichen Verlauf überprüfen und bestätigen:
- laborinterne Reproduzierbarkeit; muss regelmäßig verifiziert werden, insbesondere in der Nähe der Cutoff-
Werte, und muss bei jeder neuen Reagenzcharge neu bewertet werden - Genauigkeit (unter Verwendung von Referenzmaterial)
- Verifizierung (und ggf. Anpassung) von Referenzwerten für die Laborpopulation
Diskussion
Bei den zu validierenden Parametern und den entsprechenden Methoden, die in diesem Whitepaper genannt werden, handelt es sich lediglich um Beispiele und nicht um eine vollständige Liste. Jedes Labor muss selbst festlegen, welche Parameter im Kontext seiner IVD-Versuche und der für das Labor relevanten Normen zu validieren sind, und es muss ein optimales Protokoll zum Erreichen dieser Zielsetzungen definieren.
Die ISO 15189 gibt nicht vor, wie eine bestimmte Anforderung oder Bestimmung umzusetzen ist, rät jedoch mit dem Ziel einer ständigen Verbesserung zu einem wirksamen QMS, das alle Betriebsteile integriert.
Während die Anforderungen der ISO 15189 für alle IVD-Verfahren gleich sind, gehört die Durchflusszytometrie sicherlich zu denjenigen, bei denen sich die Akkreditierung – vor allem bei Einsatz von LDTs – am schwierigsten gestaltet.10 Die Akkreditierungsregeln wurden für hochstandardisierte Testsysteme geschaffen. Im Gegensatz zu anderen Fachgebieten wie der Hämatologie oder der klinischen Chemie, die stark standardisiert sind und auf automatisierten und gebrauchsfertigen, handelsüblichen Assays beruhen, weisen Mehrfarben-Panels für die Durchflusszytometrie
zusätzliche Komplexitätsschichten auf (z. B. Assay-Entwicklung, manuelle Probenvorbereitung, komplexe Datenverarbeitung/ ‑analyse).
Zudem sind Diagnose und Verlaufskontrolle von hämatologischen Malignomen komplex und dynamisch. Die Testprotokolle werden ständig im Licht der neuesten Stellungnahmen/Daten überprüft, insbesondere dann, wenn neue Marker, Farbstoffe, Geräte und Softwareanwendungen auf den Markt kommen, mit denen mehr Parameter analysiert werden können. Die Akkreditierung solcher Methoden nach der ISO 15189 erfordert einen erheblichen Aufwand und sorgfältige Planung; hinzu kommt, dass neue Anforderungen der IVDR, wie die in Anhang I aufgeführten, den Einsatz
von LDTs in Europa auf solche Anwendungen beschränken könnten, bei denen sie unerlässlich sind, weil aktuell keine Alternativen verfügbar sind.
Die ISO 15189 und die IVDR legen die Messlatte für LDTs höher. Daher werden klinische Labore wahrscheinlich nur noch dann auf LDTs zurückgreifen, wenn es keine Alternativen gibt, und ansonsten entweder zu im Handel erhältlichen IVDAssays übergehen oder für ihre LDTs Komponenten, die bereits die CE-IVD-Kennzeichnung tragen, gemäß Herstelleranweisung
einsetzen.
Durch die IVDR verschärfen sich die Qualitäts- und Validierungsanforderungen für Hersteller beträchtlich. Somit liegt es auf der Hand, dass dieselben IVD-Tests, wenn sie in Laboren entwickelt werden, ebenfalls genauer kontrolliert werden. Dieser Trend zu einer stärkeren Kontrolle von LDTs ist nicht auf Europa beschränkt. In den USA beispielsweise erwägt auch die Food and Drug Administration (FDA) eine Regulierung von LDTs.11 Sie hat bereits zu verstehen gegeben, dass sie es für problematisch hält, wenn ein von einem IVD-Hersteller entwickelter Test anders reguliert würde als ein identischer laborentwickelter Test. 12
Quellen
- The NHS, National Pathology Program, https://www.england.nhs.uk/wp-content/uploads/2014/02/pathol-dig-first.pdf
- International Organization for Standardization (ISO) 15189. Schneider et al. Ann Lab Med. 2017 Sep
- ISO 15189:2012 Medical laboratories - Requirements for quality and competence. Westgard QC. Pereira, P. (February 2017).
- Medical biology in the face of the evolution of health care needs. Académie Nationale de Pharmacie. 2018
- Laboratoires de biologie médicale : analyse des coûts liés à l’accréditation selon la norme EN ISO 15189. Syndicat National des Médecins Biologistes. Mai 2011. http://www.bioprat.com/modules/upload/upload/coutsaccreditation.pdf
- SH-GTA 04 technical guide, https://tools.cofrac.fr/documentation/SH-GTA-04
- ISO 5725-4:1994. Accuracy (trueness and precision) of measurement methods and results -- Part 4, https://www.iso.org/fr/standard/11836.html
- Flow cytometry and the stability of phycoerythrin-tandem dye conjugates. R. Hulspas et al. Cytometry Part A. 2009
- SH FORM 43, COFRAC. https://tools.cofrac.fr/fr/documentation/index.php?fol_id=64
- Accreditation of Flow Cytometry in Europe. Sack et al. Cytometry Part B (Clinical Cytometry) 84B:135–142 (2013)
- FDA Notification and Medical Device Reporting for Laboratory Developed Tests (LDTs), Draft Guidance Document, US FDA, 2014 https://www.fda.gov/regulatory-information/search-fda-guidance-documents/fda-notification-and-medical-device-reporting-laboratory-developed-tests-ldts
- Discussion Paper on Laboratory Developed Tests (LDTs), US FDA, 2017 https://www.fda.gov/media/102367/download
